
Galactosemia
Overview
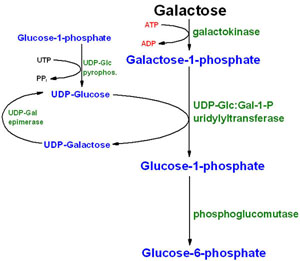
As shown in the table below, 3 types of galactosemia can occur.
| Enzyme Deficiency | Differentiating Characteristics |
|---|---|
| Galactose-1-phosphate uridyl-transferase (GALT) |
Results in classic galactosemia, in which homozygotes have
absent or barely detectable enzyme activity. If untreated, severe
feeding difficulties, failure to thrive, and liver failure can occur
within the first few days of life and lead to death if not managed
appropriately. Children with Duarte variant galactosemia generally have a residual 25% GALT enzyme activity of approximately 25%. It occurs when a child inherits a classic galactosemia mutation from one parent and the Duarte variant from the other. Once previously thought to cause a milder type of classic galactosemia, it has since been shown to have no measurable clinical impact when left untreated. |
| Galactokinase 1 (GALK1) | Results in early cataract development that is reversible if treated appropriately. Occasionally may also cause increased cranial pressure, but individuals are otherwise asymptomatic. |
| UDP-galactose-4-epimerase (GALE) | Can be subtyped as a generalized deficiency in which all tissues are affected and can result in similar signs/symptoms of classic galactosemia. Alternatively, those with peripheral or intermediate GALE deficiency are generally asymptomatic and do not require therapy. |
GALT deficiency causes the most common and severe form of galactosemia and will be the focus of the following discussion.
Other Names & Coding
E74.21, Galactosemia
Further coding details can be found at ICD-10 for Galactosemia (icd10data.com).
Prevalence
GALK deficiency seems to be rare; although there have been no large population studies, the estimated prevalence is less than 1:100,000. [Berry: 2021]
GALE deficiency in its generalized symptomatic form is exceedingly rare, with only a few reported patients ever identified in the United States. The asymptomatic peripheral and intermediate forms are much more common with estimates of 1:6,700 newborn African Americans and 1:70,000 newborn Caucasian Americans. [Alano: 1997] [Fridovich-Keil: 2008]
Genetics
Prognosis
Practice Guidelines
Specific guidelines for the diagnosis, treatment, and follow-up
have been established by an international consortium of specialists and the
Galactosemia Network as follows:
Welling L, Bernstein LE, Berry GT, Burlina AB, Eyskens F, Gautschi M, Grünewald S, Gubbels CS, Knerr I, Labrune P, van der
Lee JH, MacDonald A, Murphy E, Portnoi PA, Õunap K, Potter NL, Rubio-Gozalbo ME, Spencer JB, Timmers I, Treacy EP, Van Calcar
SC, Waisbren SE, Bosch AM.
International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up.
J Inherit Metab Dis.
2017;40(2):171-176.
PubMed abstract / Full Text
Roles of the Medical Home
Clinical Assessment
Pearls & Alerts for Assessment
Sepsis in newborns may signal galactosemiaEscherichia coli is the most common bacteria causing sepsis in infants with galactosemia, but Klebsiella, Enterobacter, Staphylococcus, group-B strep, and Streptococcus faecalis also have been observed.
Osteoporosis common in children with galactosemiaEvaluation of calcium intake while avoiding galactose rich foods is necessary to optimize bone health. Regular assessments of vitamin D levels and supplementation, if necessary, are recommended as well. Surveillance with DEXA scanning is recommended at six years of age, during puberty, through adolescence, and every 5 years as an adult. [Berry: 2021]
High false positive newborn screen resultsMost children who have a positive newborn screening test will ultimately be found to carriers of classic galactosemia or Duarte galactosemia. Nonetheless, a newborn with questionable screening results should be treated with soy-based formula pending definitive results from further tests.
Screening
For the Condition
Of Family Members
For Complications
- Delays in speech and language development
- Delays in cognitive or learning development
- Cataracts (though they may have little impact on vision) [Widger: 2010]
- Sudden increases in toxic analytes – RBC gal-1-P and urinary galactitol, generally monitored through periodic visits with metabolic genetics
- Plasma 17-beta-estradiol and FSH in girls suspected to have primary ovarian insufficiency/failure [Welling: 2017]
Presentations
- Jaundice
- Vomiting
- Hepatomegaly
- Failure to thrive
- Poor feeding
- Lethargy and Irritability
- Diarrhea
- Sepsis (usually Escherichia coli sepsis)
- Poor growth
- Learning disabilities
- Speech apraxia
- Unsteady gait
- Developmental delays
- Cataracts (reversible with dietary treatment)
- Decreased bone mineral density
- Premature ovarian insufficiency
- Ataxia and tremor
The Q188R and K285N variants common in European populations are generally associated with more severe disease and risk for complication. [Berry: 2021]
Clinical Classification
Differential Diagnosis
- Biliary atresia [Müller: 1997]
- Fanconi-Bickel syndrome [Sakura: 2000]
- Gestational alloimmune liver disease
- Portosystemic vascular shunts
History & Examination
Current & Past Medical History
Developmental & Educational Progress
Maturationalprogress
Physical Exam
Testing
Genetic Testing
Specialty Collaborations & Other Services
Biochemical Genetics (Metabolics) (see NW providers [1])
Nutrition, Metabolic (see NW providers [11])
Treatment & Management
Overview
Pearls & Alerts for Treatment & Management
Complications despite dietary restrictionDespite even rigorous galactose avoidance, individuals may still have symptoms including developmental delays, mild to moderate intellectual disability, growth problems, speech and language problems, ataxia, and tremors. Additionally, many women will develop premature ovarian insufficiency.
Hidden lactose in foodsLactose may be found in whey, milk solids, and dry milk powder, as well as in some non-milk products like fermented soy, legumes, tomato sauces, and organ meats.
Hidden lactose in medicationCertain medications have galactose or lactose fillers. These are not required to be listed in supplements. Avoid casein hydrolysates (Alimentum®, Nutramigen®, Pregestimil®) and medications with lactulose. [Berry: 2021]
Fruit and vegetable galactose controversyFree galactose is present in some fruits and vegetables, such as tomatoes, Brussels sprouts, bananas, and apples. There is no consensus about whether these should be eliminated from the diet because endogenous synthesis of galactose also occurs. Some have suggested that an elemental formula (galactose-free) may be preferable to soy formula in the treatment of galactosemia. [Gropper: 2000] [Segal: 1998] [Zlatunich: 2005]
Systems
Genetics
Specialty Collaborations & Other Services
Biochemical Genetics (Metabolics) (see NW providers [1])
Genetic Testing and Counseling (see NW providers [5])
Development (general)
Specialty Collaborations & Other Services
Early Intervention for Children with Disabilities/Delays (see NW providers [3])
Developmental - Behavioral Pediatrics (see NW providers [1])
Physical Therapy (see NW providers [0])
Occupational Therapy (see NW providers [1])
Speech - Language Pathologists (see NW providers [4])
Eyes/Vision
Specialty Collaborations & Other Services
Pediatric Ophthalmology (see NW providers [1])
Endocrine/Metabolism
Specialty Collaborations & Other Services
Pediatric Endocrinology (see NW providers [1])
Ask the Specialist
An older sibling had a false positive NBS for galactosemia, so do I still need to put the baby on soy formula?
Yes, all newborns with an abnormal NBS, regardless of symptoms or family history, should be placed on soy formula until confirmatory testing has resolved whether the newborn is actually affected.
Should siblings of an affect child be tested for galactosemia?
For classic galactosemia, if the siblings were born in the USA, they would have been screened for the condition already by the state Newborn Screen and if normal, are not at risk. However, they may be carriers for the condition, which would be important to know when they are old enough to have children. Genetic testing for carrier status is not recommended for children until they are adults and can make their own reproductive decisions.
How does galactosemia differ from lactose intolerance?
Persons with galactosemia are unable to produce the GALT enzyme, and therefore are unable to fully metabolize galactose and lactose. This causes a toxic build-up of other metabolites that cause significant liver dysfunction and damage. Alternatively, lactose intolerance is a deficiency of the enzyme lactase produced by the intestinal tract, which results in intestinal gas, bloating, and diarrhea when exposed to lactose. Though there are similarities in the restricted foods, persons with lactose intolerance may be able to tolerate small amounts of lactose safely and are not generally at risk for serious health consequences if exposed. Additionally, persons with lactose intolerance may take lactase medications orally to help digest the lactose, which does not resolve the defect in galactosemia patients.
Resources for Clinicians
On the Web
The Medical Home Portal's newborn disorders page for galactosemia provides information about the newborn screening process, follow-up on positive screening results, and steps to be taken after diagnosis confirmation.
Galactosemia (GeneReviews)
Detailed information addressing clinical characteristics, diagnosis/testing, management, genetic counseling, and molecular
pathogenesis; from the University of Washington and the National Library of Medicine.
Classic Galactosemia (Orphanet)
Overview of galactosemia and links to more information, services, and other resources; from Orphanet, a French-coordinated
consortium involving over 40 countries to provide a portal for information about rare diseases and orphan drugs.
Helpful Articles
PubMed search for galactosemia and neonatal screening, last 5 years.
Welling L, Meester-Delver A, Derks TG, Janssen MCH, Hollak CEM, de Vries M, Bosch AM.
The need for additional care in patients with classical galactosaemia.
Disabil Rehabil.
2019;41(22):2663-2668.
PubMed abstract
Rubio-Gozalbo ME, Haskovic M, Bosch AM, Burnyte B, Coelho AI, Cassiman D, Couce ML, Dawson C, Demirbas D, Derks T, Eyskens
F, Forga MT, Grunewald S, Häberle J, Hochuli M, Hubert A, Huidekoper HH, Janeiro P, Kotzka J, Knerr I, Labrune P, Landau YE,
Langendonk JG, Möslinger D, Müller-Wieland D, Murphy E, Õunap K, Ramadza D, Rivera IA, Scholl-Buergi S, Stepien KM, Thijs
A, Tran C, Vara R, Visser G, Vos R, de Vries M, Waisbren SE, Welsink-Karssies MM, Wortmann SB, Gautschi M, Treacy EP, Berry
GT.
The natural history of classic galactosemia: lessons from the GalNet registry.
Orphanet J Rare Dis.
2019;14(1):86.
PubMed abstract / Full Text
Clinical Tools
Care Processes & Protocols
ACT Sheet for Classical Galactosemia (ACMG)
Provides recommendations for clinical and laboratory follow-up of the newborn with out-of-range screening results, along with
national and local resources for clinicians and families; American College of Medical Genetics.
ACT Sheet for Primary or Secondary Hypergalactosemia (ACMG)
Contains short-term recommendations for clinical follow-up of the newborn who has screened positive; American College of Medical
Genetics.
Resources for Patients & Families
Information on the Web
Galactosemia (MedlinePlus)
Information for families includes a description, frequency, causes, inheritance, other names, and additional resources; from
the National Library of Medicine.
Galactosemia (GARD)
Includes information about symptoms, inheritance, diagnosis, finding a specialist, related diseases, and support organizations;
Genetic and Rare Diseases Information Center of the National Center for Advancing Translational Sciences.
Resources for Galactosemia (Disease InfoSearch)
Compilation of information, articles, research, case studies, and genetics links; from Genetic Alliance.
National & Local Support
Galactosemia Foundation
Provides information about galactosemia and facilitates networking among families, clinicians, and researchers.
Studies/Registries
Studies of Galactosemia (clinicaltrials.gov)
Studies looking at better understanding, diagnosing, and treating this condition; from the National Library of Medicine.
Services for Patients & Families Nationwide (NW)
| Service Categories | # of providers* in: | NW | Partner states (4) (show) | | NM | NV | RI | UT | |
|---|---|---|---|---|---|---|---|---|---|
| Biochemical Genetics (Metabolics) | 1 | 1 | 2 | 3 | 2 | ||||
| Developmental - Behavioral Pediatrics | 1 | 2 | 3 | 12 | 9 | ||||
| Early Intervention for Children with Disabilities/Delays | 3 | 34 | 30 | 13 | 51 | ||||
| Genetic Testing and Counseling | 5 | 5 | 11 | 7 | 10 | ||||
| Nutrition, Metabolic | 11 | 11 | 13 | 13 | 11 | ||||
| Occupational Therapy | 1 | 17 | 22 | 22 | 37 | ||||
| Pediatric Endocrinology | 1 | 4 | 6 | 12 | 7 | ||||
| Pediatric Ophthalmology | 1 | 6 | 6 | 8 | 4 | ||||
| Physical Therapy | 12 | 9 | 7 | 40 | |||||
| Speech - Language Pathologists | 4 | 23 | 11 | 34 | 65 | ||||
For services not listed above, browse our Services categories or search our database.
* number of provider listings may vary by how states categorize services, whether providers are listed by organization or individual, how services are organized in the state, and other factors; Nationwide (NW) providers are generally limited to web-based services, provider locator services, and organizations that serve children from across the nation.
Bibliography
Alano A, Almashanu S, Maceratesi P, Reichardt J, Panny S, Cowan TM.
UDP-galactose-4-epimerase deficiency among African-Americans: evidence for multiple alleles.
J Invest Med.
1997;45:191A.
Berry GT.
Classic Galactosemia and Clinical Variant Galactosemia.
GeneReviews.
2021.
PubMed abstract / Full Text
Berry GT.
Galactosemia: when is it a newborn screening emergency?.
Mol Genet Metab.
2012;106(1):7-11.
PubMed abstract
An excellent review regarding response to, and family counseling for, children who have a positive newborn screening result
for galactosemia.
Bosch AM, Maurice-Stam H, Wijburg FA, Grootenhuis MA.
Remarkable differences: the course of life of young adults with galactosaemia and PKU.
J Inherit Metab Dis.
2009;32(6):706-12.
PubMed abstract
Investigates the course of life and the social demographical outcomes in young adults with galactosaemia and compares them
with the general population and with PKU patients.
Elsas LJ.
Galactosemia.
GeneReview.
2010.
PubMed abstract / Full Text
Includes disease characteristics and genetic, diagnosis, and management information.
Fridovich-Keil JL, Walter JH.
Galactosemia. In: Valle D, Beaudet A, Vogelstein B, Kinzler K, Antonarakis S, Ballabio A, eds. The Online Metabolic and Molecular
Bases of Inherited Disease (OMMBID). Chap 72.
McGraw-Hill;
2008.
Gropper S.
Free Galactose Content of Fresh Fruits and Strained Fruit and Vegetable Baby Foods: More Foods to Consider for the Galactose-restricted
Diet.
Journal of the American Dietetic Association.
2000;100(5).
PubMed abstract
Examines free galactose content of foods and discusses restriction issues.
Hoffmann B, Dragano N, Schweitzer-Krantz S.
Living situation, occupation and health-related quality of life in adult patients with classic galactosemia.
J Inherit Metab Dis.
2012.
PubMed abstract
Evaluates psychosocial, educational, and occupational outcome as well as health-related quality of life in adult German patients
with galactosemia. Compares information with data from patients with phenylketonuria as well as the general German population.
McCorvie TJ, Timson DJ.
Structural and molecular biology of type I galactosemia: disease-associated mutations.
IUBMB Life.
2011;63(11):949-54.
PubMed abstract
Michael Woods.
Galactosemia.
EBSCO Publishing; (2014)
Westside Regional Medical Center website.
Müller D, Santer R, Krawinkel M, Christiansen B, Schaub J.
Fanconi-Bickel syndrome presenting in neonatal screening for galactosaemia.
J Inherit Metab Dis.
1997;20(4):607-8.
PubMed abstract
Rubio-Gozalbo ME, Haskovic M, Bosch AM, Burnyte B, Coelho AI, Cassiman D, Couce ML, Dawson C, Demirbas D, Derks T, Eyskens
F, Forga MT, Grunewald S, Häberle J, Hochuli M, Hubert A, Huidekoper HH, Janeiro P, Kotzka J, Knerr I, Labrune P, Landau YE,
Langendonk JG, Möslinger D, Müller-Wieland D, Murphy E, Õunap K, Ramadza D, Rivera IA, Scholl-Buergi S, Stepien KM, Thijs
A, Tran C, Vara R, Visser G, Vos R, de Vries M, Waisbren SE, Welsink-Karssies MM, Wortmann SB, Gautschi M, Treacy EP, Berry
GT.
The natural history of classic galactosemia: lessons from the GalNet registry.
Orphanet J Rare Dis.
2019;14(1):86.
PubMed abstract / Full Text
Sakura N, Mizoguchi N, Ono H, Yamaoka H, Hamakawa M.
Congenital biliary atresia detected as a result of galactosemia screening by the Beutler method.
Clin Chim Acta.
2000;298(1-2):175-9.
PubMed abstract
Segal S. .
Galactosaemia today: the enigma and the challenge - Komrower Lecture.
J Inherit Metab Dis.. 1998; (21):455-471. Netherlands.: SSIEM and Klower Academic Publishers; http://link.springer.com/article/10.1023%2FA%3A1005402618384#page-1
Presents the challenges of managing galactosemia.
Stuhrman G, Perez Juanazo SJ, Crivelly K, Smith J, Andersson H, Morava E.
False-Positive Newborn Screen Using the Beutler Spot Assay for Galactosemia in Glucose-6-Phosphate Dehydrogenase Deficiency.
JIMD Rep.
2017;36:1-5.
PubMed abstract / Full Text
Therrell BL, Padilla CD, Loeber JG, Kneisser I, Saadallah A, Borrajo GJ, Adams J.
Current status of newborn screening worldwide: 2015.
Semin Perinatol. 2015 Apr;39(3):171-187.; (2015)
https://pubmed.ncbi.nlm.nih.gov/25979780/. Accessed on 6/28/2021.
Welling L, Bernstein LE, Berry GT, Burlina AB, Eyskens F, Gautschi M, Grünewald S, Gubbels CS, Knerr I, Labrune P, van der
Lee JH, MacDonald A, Murphy E, Portnoi PA, Õunap K, Potter NL, Rubio-Gozalbo ME, Spencer JB, Timmers I, Treacy EP, Van Calcar
SC, Waisbren SE, Bosch AM.
International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up.
J Inherit Metab Dis.
2017;40(2):171-176.
PubMed abstract / Full Text
Welling L, Meester-Delver A, Derks TG, Janssen MCH, Hollak CEM, de Vries M, Bosch AM.
The need for additional care in patients with classical galactosaemia.
Disabil Rehabil.
2019;41(22):2663-2668.
PubMed abstract
Widger J, O'Toole J, Geoghegan O, O'Keefe M, Manning R.
Diet and visually significant cataracts in galactosaemia: is regular follow up necessary?.
J Inherit Metab Dis.
2010;33(2):129-32.
PubMed abstract
Zlatunich CO, Packman S.
Galactosaemia: early treatment with an elemental formula.
J Inherit Metab Dis.
2005;28(2):163-8.
PubMed abstract